
Polymerase Chain Reaction (PCR) is one of the most transformative techniques in modern biology. Since its development in the 1980s by Kary Mullis, it has become a cornerstone of genetic research, forensic science, medical diagnostics, and biotechnology. At the heart of PCR lies a fundamental question: why does it rely on DNA rather than RNA? The answer involves chemistry, stability, enzyme compatibility, and practical application. Understanding this distinction is essential for anyone working in molecular biology or interpreting diagnostic results.
The Chemical Nature of DNA vs. RNA
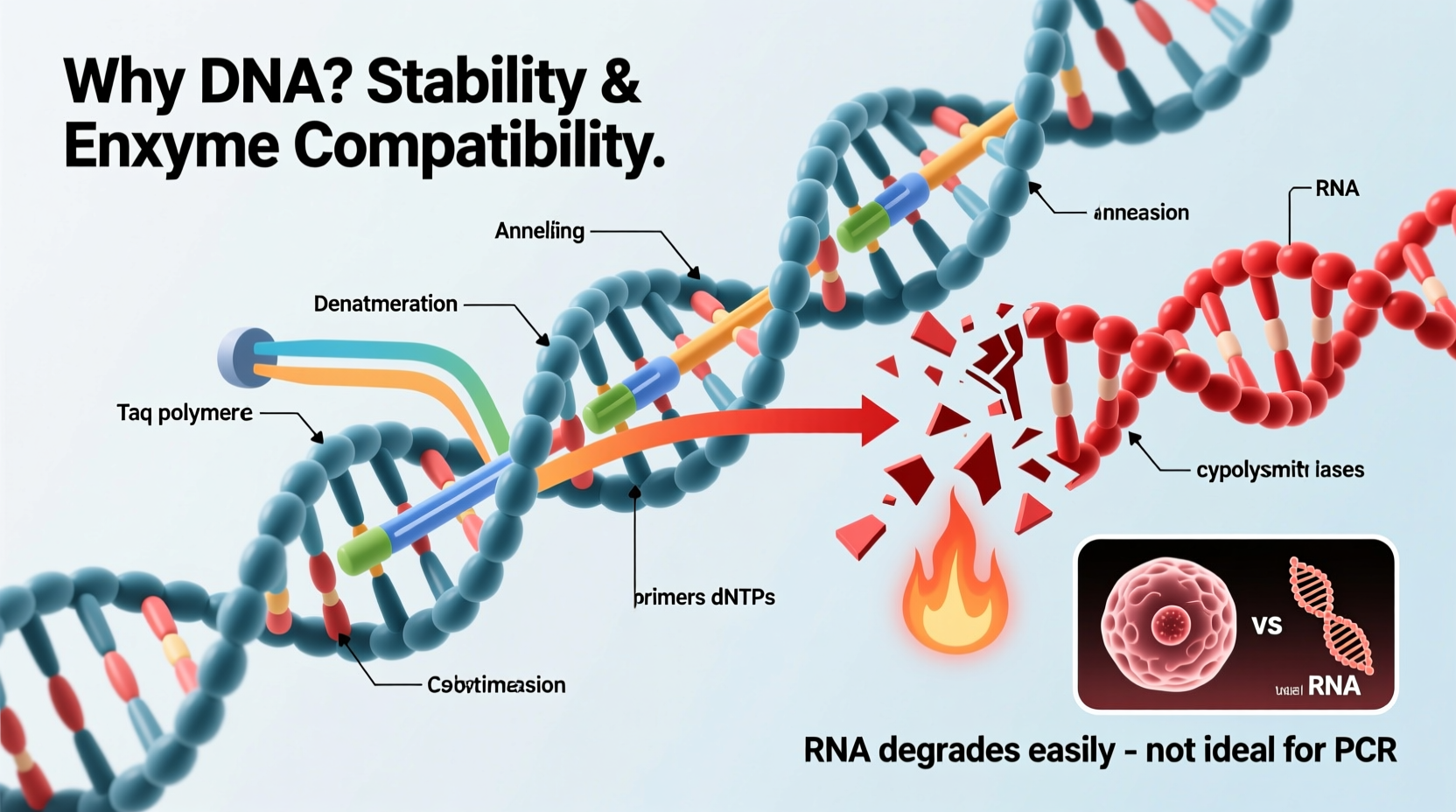
DNA and RNA are both nucleic acids that store and transmit genetic information, but their chemical structures differ in ways that make DNA far more suitable for PCR.
DNA contains deoxyribose sugar, while RNA contains ribose sugar. The key difference lies in the 2’ carbon of the sugar ring: deoxyribose lacks an oxygen atom that ribose possesses. This seemingly small change has profound consequences. The extra hydroxyl group (-OH) on ribose makes RNA chemically unstable and highly susceptible to hydrolysis, especially under alkaline conditions or elevated temperatures. In contrast, DNA’s deoxyribose structure resists breakdown, making it much more durable during repeated heating cycles.
PCR involves repeated temperature changes—denaturation at around 95°C, annealing at 50–65°C, and extension at 72°C. These cycles can span 30 to 40 repetitions. RNA would degrade rapidly under such conditions, compromising the integrity of the target sequence. DNA, with its inherent thermal stability, survives these fluctuations without significant damage.
Enzyme Compatibility: Why Taq Polymerase Prefers DNA
The enzyme responsible for synthesizing new DNA strands during PCR—typically Taq polymerase—is a DNA-dependent DNA polymerase. This means it reads a DNA template and builds a complementary DNA strand. It cannot use RNA as a template.
In contrast, enzymes that replicate RNA—such as reverse transcriptase—are specialized and function differently. They are RNA-dependent DNA polymerases, capable of converting RNA into complementary DNA (cDNA). But this is not part of standard PCR; it's a prerequisite step for a variation called RT-PCR (Reverse Transcription PCR).
Taq polymerase was isolated from *Thermus aquaticus*, a bacterium that thrives in hot springs. Its heat resistance allows it to remain active through multiple denaturation cycles. However, no naturally occurring thermostable enzyme efficiently copies RNA to RNA or RNA to DNA across dozens of cycles under PCR conditions. Thus, the enzymatic machinery of conventional PCR is fundamentally designed for DNA amplification.
“PCR’s power comes from its simplicity and specificity—but only because it works within the stable framework of double-stranded DNA.” — Dr. Laura Simmons, Molecular Biologist, NIH
Stability and Practicality in Sample Handling
Another major reason PCR uses DNA is practicality. DNA is robust and can be stored for long periods at -20°C or even room temperature when dried or stabilized. Blood spots on filter paper, buccal swabs, and preserved tissue samples all retain usable DNA for months or years.
RNA, on the other hand, is notoriously fragile. It is rapidly degraded by ubiquitous enzymes called RNases, which are present on skin, in dust, and on laboratory surfaces. Even brief exposure can destroy an RNA sample. This fragility makes RNA impractical for routine PCR unless special precautions are taken—and even then, it's treated as a temporary intermediate.
In clinical settings, where speed and reliability are paramount, using DNA reduces the risk of false negatives due to sample degradation. Forensic labs, for example, often work with old or compromised biological evidence. DNA’s resilience ensures that usable genetic material can still be amplified even from decades-old samples.
Step-by-Step: How PCR Leverages DNA Structure
The success of PCR hinges on a precise sequence of events, all optimized for DNA:
- Denaturation (95°C): Double-stranded DNA is heated to separate it into two single strands. The hydrogen bonds between base pairs break, but the covalent backbone of DNA remains intact.
- Annealing (50–65°C): Sequence-specific primers bind to complementary regions flanking the target DNA segment. These primers are short synthetic DNA strands designed to match the ends of the region of interest.
- Extension (72°C): Taq polymerase adds nucleotides to the 3’ end of each primer, synthesizing a new DNA strand complementary to the template.
- Repetition: Steps 1–3 repeat, doubling the number of DNA copies each cycle. After 30 cycles, over a billion copies of the original sequence can be produced.
This entire process relies on the predictable behavior of double-stranded DNA. RNA typically exists as a single strand and forms complex secondary structures (like hairpins), which interfere with primer binding and polymerase progression. Even if RNA could survive the heat, its structural unpredictability would reduce amplification efficiency and specificity.
When RNA Is Used: The Role of RT-PCR
While standard PCR uses DNA, there are cases where RNA is the molecule of interest—particularly in gene expression studies or viral detection (e.g., SARS-CoV-2 testing). In these instances, scientists use **RT-PCR**, which begins with reverse transcription.
In RT-PCR, RNA is first converted into cDNA using reverse transcriptase and a primer (often oligo-dT or random hexamers). Once cDNA is generated, the sample enters the standard PCR cycle. Effectively, RT-PCR is a two-step process: RNA → cDNA → amplified DNA.
This hybrid approach allows researchers to study RNA-based phenomena while still benefiting from the robustness and scalability of DNA amplification. But critically, the actual PCR phase still operates on DNA—not RNA.
| Feature | DNA in PCR | RNA in Standard PCR |
|---|---|---|
| Thermal Stability | High – withstands 95°C cycles | Low – degrades quickly at high temps |
| Enzyme Compatibility | Fully compatible with Taq polymerase | Not recognized by DNA polymerases |
| Sample Longevity | Years when properly stored | Minutes to hours without protection |
| Natural Form | Double-stranded, predictable | Single-stranded, structured |
| Amplification Efficiency | High and reproducible | Unreliable without conversion |
Mini Case Study: Detecting Viral Infections
During the early days of the COVID-19 pandemic, diagnostic labs faced immense pressure to deliver accurate tests. The SARS-CoV-2 virus carries its genome in RNA form. To detect it, laboratories used RT-PCR.
A nasal swab collected from a patient contained viral RNA. Technicians immediately placed the sample in a stabilizing buffer to inhibit RNases. In the lab, RNA was extracted and mixed with reverse transcriptase and primers targeting specific viral genes. This produced cDNA. From that point forward, the process followed standard PCR protocols: denaturation, annealing, extension—repeated 40 times.
The final result was a measurable signal indicating whether the virus was present. Crucially, the actual amplification—the exponential copying—occurred only after RNA was converted to DNA. This case illustrates why PCR itself cannot work directly on RNA: stability and enzyme constraints demand a DNA intermediary.
Checklist: Key Reasons PCR Uses DNA Instead of RNA
- ✅ DNA is chemically stable under high temperatures used in PCR cycling
- ✅ Taq polymerase requires a DNA template to synthesize new strands
- ✅ DNA maintains a predictable double-helix structure ideal for primer binding
- ✅ DNA samples are easier to store and less prone to environmental degradation
- ✅ No widespread thermostable RNA-dependent RNA polymerase exists for routine use
- ✅ Contamination risks from RNases make RNA handling impractical for standard workflows
Frequently Asked Questions
Can PCR amplify RNA directly?
No, standard PCR cannot amplify RNA directly. RNA must first be reverse-transcribed into complementary DNA (cDNA) before entering the PCR process. This combined method is known as RT-PCR.
Why don’t we just modify PCR to use RNA?
Modifying PCR to use RNA would require entirely different enzymes, buffers, and conditions. RNA’s instability and structural complexity make reliable amplification extremely difficult. Converting RNA to DNA leverages the existing, highly optimized PCR infrastructure more efficiently.
Is DNA always available when RNA is present?
Not necessarily. In some cases, such as certain viruses (e.g., influenza, HIV), the genetic material exists only as RNA. There may be no DNA version unless reverse transcription occurs—either naturally in infected cells or artificially in the lab.
Conclusion: Embracing the Logic Behind PCR Design
The decision to use DNA rather than RNA in PCR is not arbitrary—it reflects deep biochemical logic. DNA offers unmatched stability, enzymatic compatibility, and structural predictability, all of which are essential for the precision and reproducibility that PCR demands. While RNA plays vital roles in cellular function and disease detection, it serves as a precursor in molecular workflows, not the final substrate for amplification.
Understanding this distinction empowers researchers, clinicians, and students to design better experiments, interpret test results accurately, and appreciate the elegance of a technique that turns a tiny fragment of DNA into a detectable signal. As new technologies emerge, including isothermal amplification and CRISPR-based detection, the foundational principles of nucleic acid stability and enzyme specificity will continue to guide innovation.








 浙公网安备
33010002000092号
浙公网安备
33010002000092号 浙B2-20120091-4
浙B2-20120091-4
Comments
No comments yet. Why don't you start the discussion?